Site pages
Current course
Participants
General
Module 1. Dairy Development in India
Module 2. Engineering, thermal and chemical proper...
Module 3. Unit operation of various dairy and food...
Module 4. Working principles of equipment for rece...
Module 5. Dairy plant design and layout, compositi...
Module 6. Deterioration in products and their cont...
Module 7. Physical, chemical and biological method...
Module 8. Changes undergone by the food components...
Module 9. Plant utilities requirement.
References
Lesson 18. Proximate analysis of food products
Foods are classified as animal, vegetable, and mineral, and are divided into subgroups according to their source or method of manufacture, factors which are intimately correlated with their chemical composition.
18. Introduction:
Definition: “Estimation of the main components of a food using procedures that allows a reasonably rapid and acceptable measurement of various food fractions without the need for sophisticated equipment or chemicals.”
|
Original Terminology |
Alternative Terminology |
|
Moisture |
Loss on Drying |
|
Ash |
Mineral elements |
|
Crude Fat |
Fat |
|
|
Ether extract |
|
Crude Protein |
Protein |
|
Nitrogen free extractives |
Carbohydrates |
|
|
Available Carbohydrates |
|
Crude Fibre |
Unavailable Carbohydrates |
|
|
Fibre |
|
|
Neutral Detergent Fibre |
|
|
Dietary Fibre |
|
|
Non-Starch Polysaccharides |
18.1 Moisture
Water, the simplest of all constituents of foods, is one of the great concern to producer, consumer and chemist. The weight of food has little significance unless the water content is taken into consideration. The accurate determination of moisture poses many challenges as the difficulty of separating all the water from the food sample, results in underestimation of moisture content. Whereas, harsher conditions to remove all moisture from a food may result into decomposition of the product, along with/or a loss in sample mass. Most of the methods for the estimation of water in food depends on the loss in weight on heating. An exposure to the air of the drying oven causes the oxidation of certain oils and other constituents. A weight gain of such constituents offsets the weight loss due to moisture. To remove this error, the drying should be performed in vacuum. The loss in weight on heating is not entirely because of water but also due to small amount of volatile substance evident to smell present in foods. Most of the spices however contain notable quantities of volatile oil that pass off with the water.
Analytical methods of moisture determination can be classified in two ways:
A) Direct methods: Moisture analysis normally involves removing water from the food samples by drying, distillation, extraction and measuring its quantity by weighing, titration and so forth, e.g., oven drying, vacuum drying, freeze drying, distillation method, Karl Fischer method, chemical desiccation, thermo-gravimetric analysis and gas chromatography.
B) Indirect methods: The indirect methods must be calibrated against standard moisture values that have been precisely determined using direct methods, e.g., refractometry, infrared absorption, near infrared reflectance spectroscopy, microwave absorption, dielectric capacitance, mass spectrometry, NMR spectroscopy, neutron scattering method, etc.
18.1.1 Air-Oven Drying Method
It is one of the most common and widely used methods for routine moisture determination. The ovens should be thermally regulated to 0.5˚C and have minimal temperature variations (<3˚C) within the oven. The main criterion of food for moisture determination by air-oven drying is that sample should be thermally stable and should not contain significant amount of volatile compounds.
18.1.2 Vacuum Oven Drying Method
It is the standard and most accurate drying method for moisture analysis of foods. The AOAC methods generally recommend that moisture content of food can be determined by heating foods at 98 to 102 0C at a pressure of 25-100 mm Hg for 2-6 h. Lower temperatures (60-70 0C) can be used for heat sensitive/ sugary food products to prevent decomposition of sugar used in products like jam, confectionery etc.
18.1.3 Distillation Method
Two types of distillation procedures exist for moisture determination;
(a) Direct distillation and (b) Reflux distillation.
18.1.3.1 Direct Distillation
In this method, a food is heated in a liquid immiscible in water and has a high boiling point (e.g., mineral oil). The water in the food like spices and herbs distils directly from this liquid, condenses and collects in a graduated tubes; the volume of the water removed is then measured.
18.1.3.2 Reflux Distillation
It makes use of the azeotropic properties of solvent mixtures. During heating, water and an immiscible solvent (toluene or xylene) distil off together at a constant ratio and frequently at a temperature lower than the boiling point of either component. For example, the boiling points of water and toluene are 100˚C and 110.6˚C, respectively, but the boiling point of the binary mixture is 85˚C; the distillation ratio of the mixture is 20% water and 80% toluene. As water is denser than toluene, the water is again collected in a suitable measuring apparatus where it separates and has its volume measured.
18.1.4 Karl Fischer Titration Method
The Karl Fischer (KF) titration has become a standard method for the moisture analysis in liquids and solids due to its selectivity, high precision and speed. The method is particularly suitable for food where heating methods give erratic results. It has been approved for dried vegetables, oils and fats, cocoa products and liquid molasses. This method is based on the quantitative reaction of water with an anhydrous solution of sulphur dioxide and iodine dissolved in pyridine and an alcohol. The Karl Fischer reagent consists of iodine, pyridine, SO2 and methanol. The titration is conducted either by volumetric method (where the end point is brown colour, determined visually) or by colorimetric titration, where the end point is determined by a potentiometer. Food samples may be directly introduced into the reaction vessel if the water is easily accessible to the reagent. In solid foods where water is not accessible, the water is frequently extracted into anhydrous methanol and then estimated.
18.1.5 Refractometry
The refractive index is a ratio of the sine of the angle of incidence made by a ray in air to the sine of the angle of refraction made by the ray in the material being tested. The refractive index increases as the moisture content decreases. By measuring the refractive index of a solution, the moisture content can be rapidly determined using an appropriate calibration curve. Refractometry is best suited for high sugar products viz., fruit products, syrups and honey. For solid or semi-solid foods, the sample can be homogenized with an anhydrous solvent (e.g., isopropanol) and then the refractive index of the solution can be measured using a refractometer. The moisture content of the sample may be calculated using the calibration curve (produced by measuring refractive index of solutions containing the same solvents with known amounts of added water) and the mass of food homogenized in the solvent.
18.2 Total Ash
Ash refers to the inorganic residue remaining after total incineration of organic matter. The ash content is an indicator of product quality and the nutritional value of food products. When a high ash figure suggests the presence of an inorganic adulterant, it is advisable to determine the acid insoluble ash.
18.2.1 Dry Ashing
Dry ashing is the most standard method for determining the ash content of a food sample. The sample is commonly ignited at 550-600°C to oxidize all organic materials without flaming. The inorganic residue that does not volatilize at that temperature is called ash. The ash content is determined from the loss of weight, which occurs from complete oxidation of sample.
18.2.2 Wet Ashing
Wet ashing is usually used for the elemental analysis. Wet ashing commonly employs concentrated nitric acid and perchloric acid or nitric acid and sulphuric acid to oxidize the organic matter of the food sample. These acids are partially removed by volatilization and the soluble minerals remain dissolved in nitric acid. Any silica present is dehydrated and made insoluble.
However, great care must be taken when using perchloric acid, because it can be explosive on contact with water.
18.2.3 Acid Insoluble Ash
The acid insoluble ash is a measure of the sandy matter and maxima are prescribed for herbs and spices. Acid insoluble ash is determined by dissolving ash in dilute hydrochloric acid (10% w/w), the liquid filtered through an ashless filter paper and thoroughly washed with hot water. The filter paper is then ignited in the original dish, cooled and weighed.
18.2.4 Sulphated Ash
This involves moistening the ash with concentrated sulphuric acid and igniting gently to constant weight. The sulphated ash gives a more reliable ash figure for sample containing varying amount of volatile inorganic substances that may be lost at the ignition temperature used.
18.3 Crude Fiber
The crude fibre representing the cell wall material left after boiling with dilute acid and alkali in the process, is a mixture of cellulose, lignin and pentosans, together with sand, silica and other mineral matter locked in the tissues and little nitrogenous matter after grinding and defatting, boiling with sulphuric acid solution, and separation and washing of the insoluble residue. This residue is boiled with sodium hydroxide solution, separated, washed, and dried and the insoluble residue is then weighed. The loss in mass on incineration is also noted.
18.4 Fat
The oils and fats from oilseeds and fruits as well as from animal fatty tissues correspond quite closely with those extracted by diethyl ether. Practically, all the sterols and phosphorus containing organic compounds notably the lecithins are extracted with the glycerides. Essential oil and resins are the chief constituents of the ether extract of certain spices. Similarly, pepper contains nitrogenous ether soluble substance, piperine (alkaloids). Other solvents viz., chloroform, carbon tetrachloride, carbon disulphide and petroleum distillates of lower or higher boiling points dissolve fats and oils and can be used but the yield and composition of the extract differ somewhat with the solvent. Free fat can be extracted by the less polar solvents such as petroleum ether and diethyl ether, whereas the bound fat requires more polar solvents viz., alcohols for their extraction. The bound fat may be broken down by hydrolysis or other chemical treatment to yield free fat. Hence, the amount of extracted fat found in food products will depend on the method of analysis used.
18.4.1 Direct Solvent Extraction Method
The free fat content can be conveniently determined in foods by extracting the dried and ground material with petroleum ether or diethyl ether in Soxhlet extraction apparatus (Fig. 18.1). Extraction in the presence of alcohols causes the release of lipoidal substances bound to proteins and carbohydrates viz., phospholipids and glycolipids. Hence, maximum extraction is obtained by a mixture of polar and non-polar solvents. This procedure co-extracts water and water soluble substances. Hence, the residue after solvent removal and the addition of anhydrous sodium sulphate needs to be extracted with petroleum ether.
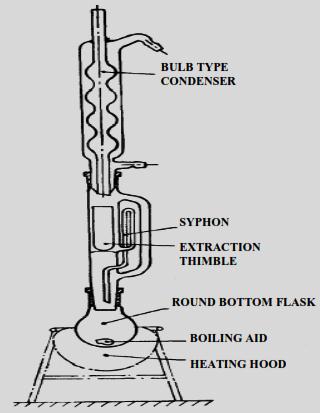
18.4.2 Solubilization Extraction Method
Bound fat can be made free if the food sample is dissolved completely prior to extraction with polar solvents. Dissolution of the food can be achieved by acid or alkaline hydrolysis. In acid hydrolysis method, the sample is heated on a steam bath with dilute HCl and boiled for 30 min. The sample solution is filtered through a wet filter paper and washed with hot water. The filter paper is then oven dried and placed directly into a Soxhlet apparatus and extracted with ethyl or petroleum ether or dichloromethane. In alkali hydrolysis method (Rose Gottlieb method), the material is treated with ammonia and alcohol in cold and the fat is extracted with diethyl ether petroleum ether mixture. The alcohol precipitates the protein, which dissolves in the ammonia; the fat can then be extracted with ether. Petroleum ether is then added as it reduces the proportion of water and hence all non-fatty substances.
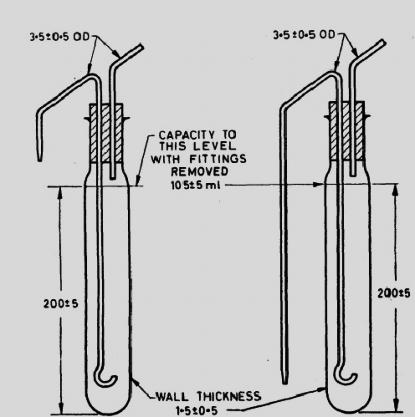
Fig. 18.2: Rose Gottlieb method
18.4.3 Volumetric Method
These involve dissolving the sample in sulphuric acid and centrifuging out the fat in specially calibrated glass vessels (butyrometers). The Gerber method is commonly employed for the routine determination of fat in milk and dairy products.
18.5 Protein
All natural foods contain protein, although trace amounts are found in honey and maple sugar. The quantification of total protein in food and food products can be performed directly or by determining total nitrogen from conversion of crude protein using a suitable conversion factor. The protein content is calculated from the total nitrogen determined by either Kjeldahl method or Dumas/Pregl-Dimas method. Amides (abundant in young shoots), ammonium salts, nitrates, lecithin, nucleic acid, purines of tea, coffee, cocoa and meat extracts in addition to protein contain nitrogen in varying proportions.
Although small, these compounds thus add error to the calculated protein estimate. However, the protein calculated by factor is a valuable figure, not only because it represents approximately the true protein present but also because it is an index of the content of other groups. The protein content can also be determined directly by formal titration, UV spectrophotometry, Lowry method, Dye binding method, IR spectrophotometry, NMR spectroscopy, turbidimetry, refractometry, etc.
18.5.1 Direct Method
Since foods contain mixtures of proteins, the methods for the direct determination of proteins need to be calibrated against a reference standard for nitrogen, e.g. Kjeldahl method.
18.5.1.1 Formal Titration Method
When formaldehyde is added to neutralized aqueous solution containing protein, the –NH2 group of protein converts to methylene-amino group (-N=CH2-) with the release of proton. This may be titrated.
18.5.1.2 Spectrophotometric Method
The Lowry method is based on the amplification of the biuret reaction
(Complex of cupric ions with protein) by subsequent reduction of the Folin phenol reagent (mixed acids of phosphomolybdic and phosphotungstic) by tyrosine and tryptophan. This redox reaction is accompanied by the formationof a blue colour (λabs745 – 750 nm), which is highly pH dependant (10-10.5).
18.5.2 Indirect Method
18.5.2.1 Kjeldahl Method
It has wide acceptance for the determination of protein in food products. The method follows three steps:
Digestion – Decomposition of organic matter by heating in the presence of concentrated sulphuric acid, the end product is ammonium sulphate solution.
Distillation – Ammonium sulphate is converted into gaseous ammonia by addition of an excess base, followed by boiling and condensation of the ammonia in a receiving solution (acid).
Titration – Quantification of the unreacted acid in the collecting vessel. The rate of digestion and the completeness of the breakdown of nitrogenous compounds to ammonium sulphate mainly depend upon the heat input, amount of boiling point elevator of acid (alkali sulphate), addition of catalyst (mercury, copper sulphate, titanium dioxide), oxidant (hydrogen peroxide), reflux rate of sulphuric acid and length of digestion. Ammonia is liberated from the acid digestion mixture by distillation in the presence of alkali (50% NaOH). A total recovery of ammonia from the digest can be obtained within 5 to 20 min by direct distillation and about 10 min by steam distillation.
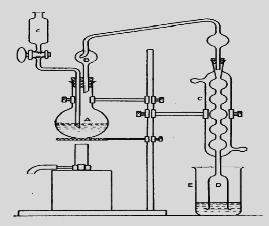
Fig. 18.3: Kjeldahl Nitrogen distillation assembly
18.5.2.2 Dumas Combustion Method
The protein content of foods can be estimated by the determination of elemental nitrogen using instruments based on the Dumas principle. In these instruments, the nitrogen containing constituents of the sample are combusted at high temperature about 1000C in the presence of oxygen to oxides of nitrogen (NOx) and then reduced over copper or tungsten to gaseous nitrogen which is measured by gas solid chromatograph using thermal conductivity detector. This method offers significant advantage over Kjeldahl method i.e., shorter analysis time (3-4 min), but these instruments appear to have limited usefulness for some food products because they can only deal with very less amount of sample.
Last modified: Thursday, 22 August 2013, 7:11 AM